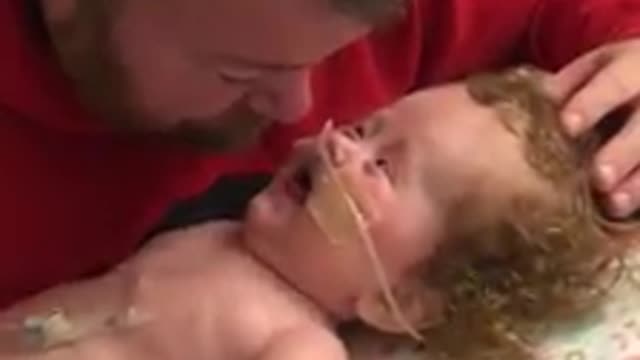
Toddler With Rare Disease Laughs With Dad And It'll Totally Melt Your Heart
This little girl named Ainsley, 2 years old, has osteogenesis imperfecta type 3. It is one of the many rare diseases that exist in the world, unfortunately. The disease results in a lack of collagen in your body, causing your bones to break or fracture easily. Despite the difficulties, Ainsley still smiles and is eager to live and enjoy every moment with his father. This is so touching!
Osteogenesis imperfecta (OI) is a heterogeneous group of hereditary diseases in which there is a disorder in the formation of type I collagen by mutations in the genes that encode that collagen. Type I collagen is a protein present in all supporting tissues, especially bone (where type I collagen is the main component of the organic matrix), skin and tendons, but also ligaments, fascias, cornea, sclera, dentin and blood vessels.
The consequences of the alteration of this collagen at bone level are the reduction of the bone matrix, with alteration of the bone structure and poor mineralization (osteopenia), so that bone resorption predominates over the formation of new bone. This implies the presence of bone fragility, frequent fractures, bone deformities and short stature. The multiple mutations described explain the great clinical heterogeneity of this pathology, existing from minimal forms to serious and lethal pictures. Other consequences of type I collagen mutation at other levels are: hyperlaxitude of ligaments and tendons, vascular fragility, decreased muscle strength, blue sclera and altered dentinogenesis.
In this case it would be osteogenesis imperfecta type 3, whose characteristics are: presence of fractures at birth and during childhood. Relative microcephaly, triangular facies, progressive bone deformities (with kyphoscoliosis, thoracic and limb deformities), chronic pain and functional disability. The final size is very low. Sclera may be blue at birth and then normalized. In general there is dentinogenesis alteration and there is no deafness. OI is a genetic disorder of autosomal dominant inheritance of connective tissue and most patients are heterozygous for a given mutation de novo in patients with no family history. Being the OI of autosomal dominant inheritance, it is surprising that the risk of recurrence for a healthy couple of having another affected child is up to 5-8%, which is too high to correspond to de novo mutations, and very low for a recessive inheritance. This is explained by the existence of a "parental mosaicism", so that in a parent can exist two different cell lines, one with the mutation of collagen type I and another without it, carrying the mutation in germinal cells and in a limited number of somatic cells, so it is asymptomatic (mosaic carrier).
The diagnosis of OI is fundamentally clinical and the classification according to Sillence is still used, based on clinical, radiological findings and type of inheritance. The findings and the severity of the picture are variable according to the type of OI. There is currently no effective, curative treatment of OI, because it cannot directly act on the formation of type I collagen. Throughout history, various medical treatments (calcitonin, anabolic steroids, etc.) have been used to try to increase bone mass without obtaining results. Treatment is currently symptomatic and should be approached in a multidisciplinary manner. The best results have been achieved with growth hormone (GH) and bisphosphonates.
-
 0:14
0:14
mommyndcity
5 years ago $16.07 earnedBaby's First Taste Of Lemon Will Melt Your Heart
14.9K -
 0:51
0:51
SWNS
6 years agoBoy with rare disease could literally be frightened to death
110 -
 0:56
0:56
WildCreatures
6 years ago $42.54 earnedGrandma And Grandson Giggle-fest Will Melt Your Heart
25K1 -
 0:23
0:23
UtahSwimAcademy
5 years ago $12.75 earnedToddler Jumps Into Pool And Swims With Ease
4.94K3 -
 1:44
1:44
JayleeMoriah
5 years ago $22.08 earnedToddler Gets Surprised On Her Birthday With New Car
43.1K3 -
 0:12
0:12
ViralHog
5 years ago $0.09 earnedStranger Danger Doesn’t Matter to a Toddler with a Dog Obsession
343 -
 1:16
1:16
SWNS
6 years agoFive-year-old with Brittle Bone Disease goes wakeboarding with dad
139 -
 1:52
1:52
SWNS
6 years agoToddler began to see terrifying monsters after disease attacked her brain
868 -
 3:06
3:06
AFV
5 years agoBest Baby Laughs
3.62K3 -
 0:12
0:12
gabregomez
6 years ago $3.55 earnedToddler gets hit with giant ball in epic slow motion
9.31K